J Biomed 2017; 2:109-116. doi:10.7150/jbm.17725 This volume Cite
Review
Endothelial Glycocalyx: Novel Insight into Atherosclerosis
Ye Zeng ![]()
Institute of Biomedical Engineering, West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, Chengdu, China.
Received 2016-9-26; Accepted 2017-5-4; Published 2017-7-17
Abstract

The evidence shows that endothelial glycocalyx exerts an important role in inflammation and atherosclerosis. We previously demonstrated that hydrodynamic force such as shear stress induces the remodeling of the major component of glycocalyx including glypican-1 with attached heparan sulfate, and that sphingosine-1-phosphate (S1P) protects the glycocalyx against syndecan-1 ectodomain shedding and induces the synthesis of HS. Actually, both shear stress and S1P imposed significant influence on inflammatory disease such as atherosclerosis. Therefore, it can be concluded that glycocalyx is a critical signaling platform for determining the fate of endothelial cells and atherosclerosis. This review integrated our current understanding of shear stress, S1P and glycocalyx, and also provided new insight/issues into the role of the glycocalyx in the process of forming and developing atherosclerosis.
Keywords: glycocalyx, sphingosine-1-phosphate, shear stress, inflammation, atherosclerosis.
Introduction
Endothelial glycocalyx mediates the endothelial mechanotransduction of shear stress and serves as a selective permeability, anti-inflammatory and anti-adhesive barrier at the luminal side of the endothelial cells (ECs) [1-3], showing a protective effect on vascular functions. The endothelial glycocalyx is reduced in atherosclerosis (AS) [4, 5]. AS is the major pathological basis causing cardiovascular and cerebrovascular diseases. Thrombosis and atherosclerotic plaque rupture lead to acute coronary syndrome, including unstable angina, acute myocardial infarction and heart attack [6-8].
In the vascular remodeling due to AS, the structural and functional adaptive changes in vessels adapt to the mechanical (such as shear stress) and chemical (such as vasoactive mediators and cytokines) microenvironments, which involve various cell activities such as cell phenotype conversion [9] and remodeling of the extracellular matrix and endothelial glycocalyx [10-14]. It is well-known that flow patterns with shear stress magnitude are changed with development of AS [15]. Shear stress induces clustering of the major components of endothelial glycocalyx including glypican-1 with attached heparan sulfate (HS) [11, 16].
S1P is a lipid mediator and mostly present in plasma that induces various cellular effects, including proliferation, differentiation, survival, and migration [17]. S1P is also emerging as a potent modulator of endothelial barrier function and vascular tone [18]. Recent studies demonstrated that sphingosine-1-phosphate (S1P) protects the endothelial glycocalyx against shedding and induces the synthesis of glycocalyx [11, 16].
Both shear stress and S1P play critical roles in modulating the structure and function of the glycocalyx, and have multiple roles in vascular homeostasis and remodeling. In the present study, we review the research progress on the structure and function of the endothelial glycocalyx, and dual effects of shear stress and S1P in AS. Finally, we conclude that the glycocalyx is a critical signaling platform for deciding the fate of ECs and vascular diseases, which may be helpful for elucidating the complicated pathological mechanism of atherosclerosis.
Structure of endothelial glycocalyx
The endothelial glycocalyx covers the apical surface of the vascular endothelial cell and directly contact with blood. The major components of the endothelial glycocalyx include glycoproteins bearing acidic oligosaccharides and terminal sialic acids (SA), and proteoglycans (PG) like heparan sulfate proteoglycans (HSPG; including syndecans and glypican-1 core proteins), and glycosaminoglycan (GAG) side chains. In the vasculature, GAGs are mainly comprised of three types, namely, heparan sulfate (HS; >50% of the total GAG pool), chondroitin sulfate (CS) and hyaluronic acid or hyaluronan (HA). HA is a kind of non-sulfating GAG, which does not connect to PG core protein, but binds with receptor CD44.
In HSPGs, there are four members in the syndecan family, including syndecan-1, -2, -3 and -4. Syndecan-1 contains five potential GAG attachment sites, three near its NH2-terminal ectodomain and two adjacent to the transmembrane domain near its COOH terminus. Only CS is found near the COOH terminus of syndecan-1 [19]. Syndecan-3 contains eight potential GAG attachment sites, five near its NH2-terminal ectodomain and three adjacent to the transmembrane domain near its COOH terminus. Syndecan-2 and 4 contains three potential GAG attachment sites near their NH2-terminal ectodomain [20, 21]. Syndecan-4 can also contain CS [22]. Glypican-1 is an extracellular glycosylphosphatidylinositol (GPI)-anchored protein, which only binds with HS. In resting conditions, syndecans mRNA and glypican-1 mRNA in human umbilical vein endothelial cells (HUVECs) are expressed in the order: syndecan-1>syndecan-4>syndecan-3>syndecan-2>glypican-1 [23].
The glycocalyx is modified under several conditions including disturbed flow exposure in large vessels [24], protease degradation [24-26], and removal of plasma components, particularly albumin [27].
Function of the endothelial glycocalyx
The glycocalyx has various functions [5, 28]. The negatively charged glycocalyx surface forms an electrostatic barrier for plasma cells and proteins, like albumin. Increased serum levels of syndecan-1 are associated with acute coagulopathy following trauma [29].
The dominant mechanism that defines widespread endothelial dysfunction is impaired expression of constitutive endothelial nitric oxide synthase (eNOS) and production of nitric oxide (NO) [30]. Knockdown of glypican-1 inhibits the activation of eNOS under shear stress [31].
Both HS-ligand binding and interactions of the PG core protein with cytoskeletal and/or signaling molecules are required for cell adhesion and migration. The endothelial alignment in flow required syndecan-1 [31] and -4 [32].
Syndecan-1, 2- and -3 might contribute to angiogenesis. Syndecan-1 plays important roles in EC survival, proliferation, and organization into capillary-like structure [33]. Shed syndecan-2 regulates angiogenesis by inhibiting EC migration via CD148 (PTPRJ) signaling [34].
A recent study demonstrated that HS is essential for the interleukin (IL)-8 induced cell migration [35]. After enzymatic removal of HS, we observed significant suppression of the IL-8-upregulated Rho GTPases including Cdc42, Rac1 and RhoA, CXCL8-increased Rac1/Rho activity, as well as the abolishment of the IL-8 induced polymerization and polarization of actin cytoskeleton and an increase in stress fibers. In cell recruitment, it has been thought that both chemokine oligomerization and binding to GAGs are required. Also, their interactions with GAGs facilitate the formation of the chemokine gradients, which provide directional cues for migrating cells [36]. Thus, the glycocalyx could be a good platform to integrate various signals.
Dual effects of shear stress on atherosclerosis
It was well known that vascular endothelial injury in the susceptible location for atherosclerosis in vessels was the prerequisite for AS formation, while the atherosclerotic plaque was the consequence of subsequent vascular repair reaction induced by shear stress [37-39]. Thus, dysfunctional endothelium in the atherosclerosis-susceptible location is an early manifestation of atherosclerosis [40].
In the atherosclerosis-susceptible location, such as branches, bifurcation, and curvatures (e.g. the aortic arch) of the arterial tree, the blood stream is subject to tremendous interference and the flow departs from pulsatile, unidirectional shear stress to create flow separation zones that include flow reversal, oscillatory shear stress and sometimes turbulence (chaotic flow) [30, 41]. In contrast, flow in adjacent undisturbed flow regions of the arteries is pulsatile and has well-defined directions.
Low shear stress induced irregularly arrangement of vascular ECs and secretion of vasoconstrictors Endothelin-1 (ET-1), Angiotensin-converting enzyme (ACE), adhesion molecules (i.e. VCAM-1, ICAM-1, P-selectin, and PECAM-1) and proinflammatory cytokines [42, 43], impaired the endothelial permeability, and facilitated the recruitment and infiltration of inflammatory cell [40]. The inflammatory monocytes further differentiate and proliferate into macrophages that ingest the oxidized low-density lipoprotein forming foam cells. The foam cells accumulate within the subintimal region to form fatty streaks. The accumulated foam cells undergo apoptosis and necrosis leading to formation of a soft, destabilizing lipid-rich necrotic core within the intima region. The abnormal proliferation and migration of the vascular smooth muscle cells was accelerated, leading to the formation of fibrous cap over the necrotic core consisting of collagen type I and III.
The AS plaque grows into the lumen of the vessel and prone to rupture, leading to gradual vascular stenosis and to form a high shear stress region [44, 45]. The features of such vulnerable plaque include greater vascularization, larger necrotic lipid core, lack of endothelial lining, and thinner fibrous cap with reduced smooth muscle density, decreased collagen content and increased inflammatory cells.
Increasing evidence suggest that high shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis [46]. High shear stress can promote the growth of the collateral vessel that has stopped growing, and increase the number of the microvessel in canine myocardial infarction region [47, 48]. In addition, high shear stress can increase the thickness of the endodermis through anti-inflammatory reaction, inhibit the cell proliferation and promote the cell apoptosis of smooth muscle cell, might induce the expression of matrix metalloproteinases (MMPs) and the degradation of extracellular matrix [46].
Therefore, it can be speculated that low shear stress induces the initial lesion, and high shear stress promotes the formation of vulnerable plaque (Fig. 1). However, high shear stress is thought to be atheroprotective as the high shear stress region (>10~15 dyn/cm2) in the normal artery is not likely to develop AS [30]. It also well known that high shear stress induces the fusiform body arrangement of vascular ECs, and secretion of active substances such as nitric oxide (NO), prostacyclin (PGI2) and superoxide dismutase (SOD), thus protecting the vessels [11, 28, 49]. It is worth to investigate why high shear stress plays distinct roles in atherosclerotic lesions and normal vessels. High shear stress induced the NO, which subsequently lead to vasodilatation, reducing shear stress [17]. In the lesion location in vessel, a continuous exposure of EC to high shear stress might induced an abnormal increase in NO production, which further induce extracellular matrix and glycocalyx degradation through MMPs [50], as well as inflammation.
Dual effects of S1P on atherosclerosis
S1P is a kind of membrane phospholipid metabolite, which is an important signal molecule in multiple cells, such as vascular EC, smooth muscle cell and fibroblast [18], and plays vital roles in cell survival, cell phenotype conversion, angiogenesis, thrombosis, wound healing, and inflammation [51]. S1P also closely associates with atherosclerosis. S1P levels are elevated in diverse tissues including liver, skeletal muscle, adipose tissue, and cardiovascular tissues, as well as plasma in high-fat diet fed mice [52].
S1P is emerging as a potent modulator of endothelial function in response to injury [18]. S1P exerts a variety of biological actions through binding with the specific G protein-coupled receptor (S1P1-5) on cell surface to activate signaling cascades or serving as a second messenger [53]. Receptor S1P1-3 prevails among all kinds of tissues in the cardiovascular system [54, 55] and has been widely investigated. S1P and its receptor S1P1 was required for embryonic angiogenesis and vascular stabilization [55]. S1P can promote the formation of actin ring around the vascular ECs and strengthen the cell-cell and cell-matrix interactions through S1P1, maintaining the permeability of the vascular wall [56]. The specific agonist of S1P1 significantly inhibits the formation and development of AS, but does not influence the S1P level in plasma [57]. By fed with a high fat diet, abnormal vascular phenotype and development of plaque was obvious in the descending aorta in the Apoe-/- and EC-specific S1PR1 null mice (S1PR1f/f VE-cadherin-Cre-ERT2), but was not in the Apoe-/- and S1PR1 wild-type mice [58]. Therefore, S1P could maintain the vascular homeostasis and prevent the development of atherosclerosis through S1PR1.
Although receptors S1P2 and S1P3 are not required for embryonic angiogenesis and vascular stabilization, they cooperate with S1P1 in the process of developing embryonic vasculature [55]. The physiological levels of S1P2 and S1P3 are lower than S1P1 in vivo. Under atherosclerotic conditions, the levels of S1P and endothelial S1P2 is markedly increased [59]. S1P impairs cell-cell communication [60] and induces increases in vascular permeability and inflammation through S1P2 [61, 62]. Feeding with high cholesterol diet, the macrophage content and inflammatory cytokines such as IL-1 and IL-18 in plasma were greatly lower in the Apoe-/- and S1PR2-/- mice than that in the Apoe-/- and S1PR2 wild-type mice [63], suggesting a proatherogenic role of S1PR2 in atherosclerosis.
Alteration of shear stress during the development of atherosclerosis. From initiation to plaque rupture: (A) After endothelial dysfunction and loss of glycocalyx in the flow separation zone where the atherosclerotic lesions formation, the barrier permeability was increased, which lead to the accumulation of low-density lipoprotein, and the increase of proinflammatory cytokines and adhesion molecules under low shear stress (Low SS). This is followed by recruitment and infiltration of inflammatory monocytes. The monocytes further differentiate and proliferate into macrophages that ingest the oxidized low-density lipoprotein forming foam cells. The foam cells accumulate within the subintimal region and form fatty streaks. (B) The accumulated foam cells undergo apoptosis and necrosis leading to formation of a soft, destabilizing lipid-rich necrotic core within the intima region. The abnormal proliferation and migration of the vascular smooth muscle cells was accelerated, leading to the formation of fibrous cap over the necrotic core. The AS plaque grows into the lumen of the vessel and prone to rupture, leading to gradual vascular stenosis and to form a high shear stress region. (C) High shear stress promotes the formation of vulnerable plaque. The continuous exposure of high shear stress increased vasoactive mediators (such as S1P) and inflammatory cytokines, leading to calcification below the necrotic core, release of protease such as MMPs and neovascularization.
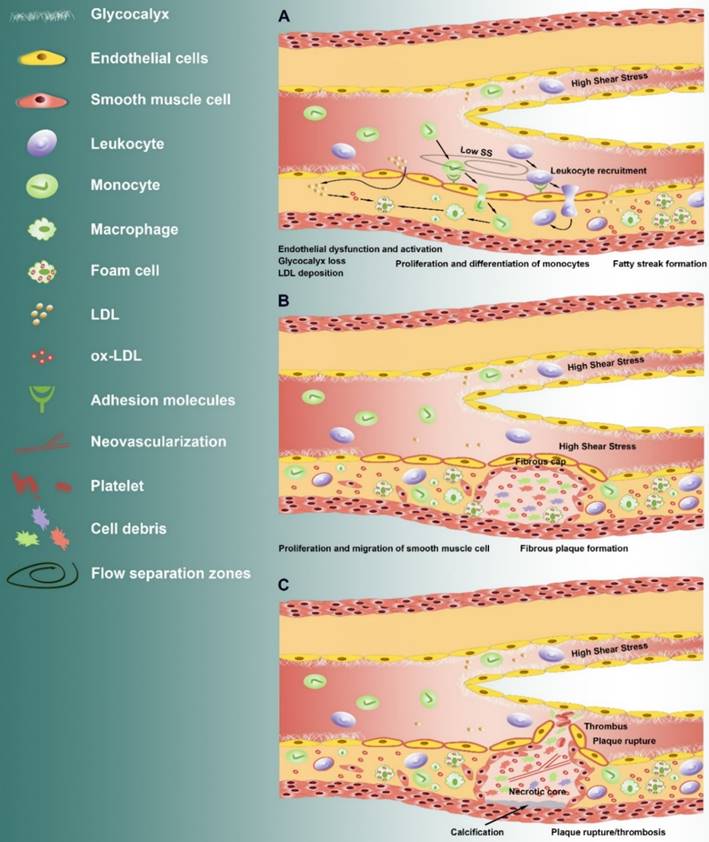
So, S1P exerts both protective role in atherosclerosis via S1P1, and proatherogenic role in atherosclerosis through S1P2 and S1P3. The maintaining of vascular permeability by S1P is in a concentration-dependent manner. What is the significance of increased S1P in atherosclerosis? How do the cells determine which receptor binds to S1P? Further investigations are needed to resolve these fundamental issues and the underlying mechanisms.
New insights on endothelial glycocalyx and atherosclerosis
Weinbaum et al. [64] pointed out that the existence of endothelial glycocalyx could weaken the shear stress on the vascular EC surface to a negligible level by theoretical analysis. Thi et al. [65] further proved that endothelial glycocalyx is required for the response of EC cytoskeleton to shear stress. Furthermore, selectively degradation of some specific components (such as HS) of the endothelial glycocalyx or silence of the specific gene (such as glypican-1) can inhibit the shear stress-induced activation of eNOS [66] and the production of NO in EC [67]. By using confocal microscopy, we discovered that 15 dyn/cm2 shear stress induces remodeling of endothelial glycocalyx [11, 16]. At initial 30 min, 15 dyn/cm2 shear stress induces the junctional clustering of HS via mobility of GPI-anchored glypican-1 in lipid rafts (rapid change). After 24 h, 15 dyn/cm2 shear stress induces the recovery of HS (adaptive remodeling), which shows similar distribution as present in the aorta of the rats and mice in vivo [68]. In recent, we detected the transcriptional expression of HSPGs (syndecan family and glypican-1) in human umbilical vein endothelial cell (HUVEC) responded to the distinct magnitudes of shear stress [23]. During the initial 0.5h of exposure, syndecan-1 mRNA was greatest upregulated by 4 dyn/cm2 of shear stress, and syndecan-4 mRNA was significantly upregulated by 10 dyn/cm2 and 15dyn/cm2. After 24h of exposure, the greatest increased HSPG mRNA was syndecan-4 under 4 dyn/cm2, and was syndecan-3 under 15 dyn/cm2. The changes of those molecules that may associate with the vascular homeostasis and endothelial dysfunction revealed the potential candidate components of the glycocalyx in response to cardiovascular diseases. The increases of syndecan-3, syndecan-4 and glypican-1 might contribute to the adaptive remodeling of glycocalyx [23].
The remodeling of glycocalyx might associated with changes in various EC function, such as proliferation, migration, adhesion, and the activation of eNOS and production of NO. Degradation of HS significantly inhibited the motility and proliferative responses of EC to shear stress [69], and greatly enhanced the adhesion of leukocyte on endothelium [70]. The NO production increased significantly within minutes under 15 dyn/cm2 shear stress [16]. Removal of glypican-1 inhibited the 15 dyn/cm2 shear stress-induced activation of eNOS, and further reduced the 4 dyn/cm2-inhibited eNOS activity. Glycocalyx could potentially be a good platform to integrate the signals (i.e. chemokine and shear stress) to slow or prevent the development of atherosclerosis by structure remodeling. Once the platform impaired, the interplay of signals as well as the involved mechanism might change.
The glycocalyx was modified after removal of plasma components, particularly albumin [27]. In recent, it was demonstrated that albumin carried S1P inhibits shedding of the syndecan-1 ectodomain via activation of S1P1 receptor [71], and thus maintains the normal vascular permeability in intact microvessels [72]. The depletion of plasma protein induces syndecan-1 shedding through MMP-mediated proteolytic cleavage close to the plasma membrane on the external face [71]. The shedding of syndecan-1 ectodomain also takes away the attached HS and CS [71]. After completely shedding of glycocalyx components (including syndecan-1 with attached HS and CS) by depletion of plasma protein, the addition of S1P induced the recovery of endothelial glycocalyx via PI3K pathway [73]. It was suggested that the stability of glycocalyx by S1P is at least partially due to the synthesis of glycocalyx. It can be conducted that S1P maintains the stability of glycocalyx through inhibiting the shedding and promoting the synthesis of glycocalyx together, thereby contributes in maintenance of normal vascular permeability [72], and in controlling the cardiovascular and immune functions [56].
The exact intracellular signaling pathway involved in the S1P preserved/induced glycocalyx is still not completely understood. The S1P1 phosphorylation-inhibited MMP activation might be mediated by a pathway involving phosphatidylinositide 3-kinase (PI3K)/Akt and Rac1. It was demonstrated that Akt-mediated phosphorylation of S1P1 receptor (T236) is indispensable for S1P-induced Rac activation, endothelial migration, and morphogenesis [74]. In addition, activation of S1P1 promotes endothelial barrier integrity, migration and survival through PI3k/Akt,, eNOS and Rac [75, 76]. However, the activation of endothelial S1PR3 and S1PR2 could counteract the anti-inflammatory actions-mediated by S1PR1-PI3K/Akt-eNOS pathway. As glycocalyx is critical for activation of eNOS, it is interesting to understand if glycocalyx is indispensable for roles of S1P in endothelial function and atherosclerosis.
Glycocalyx integrated the shear stress and S1P signals. Gαq/11 activation independent of S1P1 activation in endothelial cells (ECs). Glycocalyx might play important role in mechanotrasduction and interaction between Gαq/11 and PECAM-1. S1P protects the integrity of glycocalyx via S1P1 in ECs. Thus, the glycocalyx could potentially be a signaling platform that integrated the S1P and shear stress signals for maintaining vascular homeostasis.
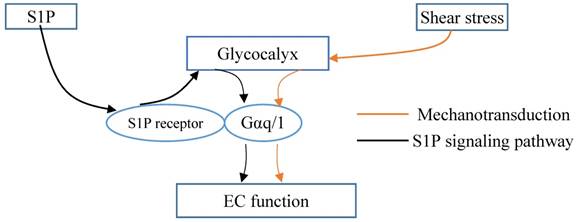
Thus, it was demonstrated the interaction between S1P1 and hemodynamics in vivo [58]. Vascular endothelial S1P1 receptor can respond to hemodynamic force, transduce the signals into the cellular promoting the stabilization of the newly formed vascular network [58]. Knockout of S1P1 gene in mouse manifests as injured vessel maturation and embryonic mortality. In areas of laminar flow with high shear stress, S1P1 is present on the membrane, whereas it is internalized in areas of disturbed flow with low shear stress. S1P1 regulates the directional migration of lymphatic endothelial cells in response to fluid shear stress, which is required plasma or S1P [77]. The change in S1P1 might associate with remodeling of the glycocalyx. However, Frangos et al. [78] reported that Gαq/11 activation independent of S1P1 activation in coronary artery endothelial cells. Glycocalyx might play important role in mechanotrasduction and interaction between Gαq/11 and PECAM-1 [79]. Therefore, the glycocalyx could potentially be a signaling platform that integrated the S1P, shear stress, chemokines and cytokines for maintaining vascular homeostasis (Fig. 2). Once the platform is destructed, the interplay among these factors and the underlying signaling pathways might be changed, which further contributes to the endothelial dysfunction and the development of atherosclerosis. Furthermore, investigations into the mechanoglycobiological mechanism underlying the remodeling of the glycocalyx could bridge the effects of shear stress and S1P on atherosclerosis. The innovations in the mechanoglycobiology field will provide new insight into developing novel prevention and treatment strategies for cardiovascular diseases.
Acknowledgements
Our work is supported by the National Natural Science Foundation of China (Grant no.11402153), and the Talent Introduction Scientific Research Projects Funded Start-Up Funds (No.2082204174089) and the Excellent Young Scientist Foundation (No. 2015SCU04A38) of Sichuan University of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tarbell JM, Ebong EE. The endothelial glycocalyx: a mechano-sensor and -transducer. Science signaling. 2008;1:pt8
2. Curry FE, Adamson RH. Endothelial glycocalyx: permeability barrier and mechanosensor. Annals of biomedical engineering. 2012;40:828-39
3. Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454:345-59
4. Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovascular research. 2010;87:300-10
5. Haeren RH, van de Ven SE, van Zandvoort MA, Vink H, van Overbeeke JJ, Hoogland G. et al. Assessment and Imaging of the Cerebrovascular Glycocalyx. Curr Neurovasc Res. 2016;13:249-60
6. Virmani R, Burke A, Farb A. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. Eur Heart J. 1998;19:678-80
7. Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276-82
8. Zeng Y. Endothelial glycocalyx as a critical signalling platform integrating the extracellular haemodynamic forces and chemical signalling. J Cell Mol Med. 2017
9. Coll-Bonfill N, de la Cruz-Thea B, Pisano MV, Musri MM. Noncoding RNAs in smooth muscle cell homeostasis: implications in phenotypic switch and vascular disorders. Pflugers Arch. 2016;468:1071-87
10. Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V. et al. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. American journal of physiology Heart and circulatory physiology. 2013;305:H811-20
11. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One. 2014;9:e86249
12. Zeng Y, Liu XH, Tarbell J, Fu B. Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Experimental cell research. 2015;339:90-5
13. Zeng Y, Ebong EE, Fu BM, Tarbell JM. The structural stability of the endothelial glycocalyx after enzymatic removal of glycosaminoglycans. PLoS One. 2012;7:e43168
14. Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. American journal of physiology Heart and circulatory physiology. 2014;306:H363-72
15. Millon A, Sigovan M, Boussel L, Mathevet JL, Louzier V, Paquet C. et al. Low WSS Induces Intimal Thickening, while Large WSS Variation and Inflammation Induce Medial Thinning, in an Animal Model of Atherosclerosis. PLoS One. 2015;10:e0141880
16. Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V. et al. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am J Physiol Heart Circ Physiol. 2013;305:H811-20
17. Tarbell JM, Simon SI, Curry FR. Mechanosensing at the vascular interface. Annu Rev Biomed Eng. 2014;16:505-32
18. Sanchez T. Sphingosine-1-Phosphate Signaling in Endothelial Disorders. Curr Atheroscler Rep. 2016;18:31
19. Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. 2006;259:339-50
20. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J. et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729-77
21. Lopes CC, Dietrich CP, Nader HB. Specific structural features of syndecans and heparan sulfate chains are needed for cell signaling. Braz J Med Biol Res. 2006;39:157-67
22. Deepa SS, Yamada S, Zako M, Goldberger O, Sugahara K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J Biol Chem. 2004;279:37368-76
23. Liu JX, Yan ZP, Zhang YY, Wu J, Liu XH, Zeng Y. Hemodynamic shear stress regulates the transcriptional expression of heparan sulfate proteoglycans in human umbilical vein endothelial cell. Cell Mol Biol (Noisy-le-grand). 2016;62:28-34
24. Brands J, Van Teeffelen JWGE, Van den Berg BM, Vink H. Role for glycocalyx perturbation in atherosclerosis development and associated microvascular dysfunction. Future Lipidology. 2007;2:527-34
25. Huxley VH, Williams DA. Role of a glycocalyx on coronary arteriole permeability to proteins: evidence from enzyme treatments. Am J Physiol Heart Circ Physiol. 2000;278:H1177-85
26. Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng. 2012;40:840-8
27. Michel CC, Phillips ME, Turner MR. The effects of native and modified bovine serum albumin on the permeability of frog mesenteric capillaries. J Physiol. 1985;360:333-46
28. Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med. 2016;280:97-113
29. Chignalia AZ, Yetimakman F, Christiaans SC, Unal S, Bayrakci B, Wagener BM. et al. The Glycocalyx and Trauma: A Review. Shock. 2016;45:338-48
30. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16-26
31. Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol (Camb). 2014;6:338-47
32. Baeyens N, Mulligan-Kehoe MJ, Corti F, Simon DD, Ross TD, Rhodes JM. et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc Natl Acad Sci U S A. 2014;111:17308-13
33. Lamorte S, Ferrero S, Aschero S, Monitillo L, Bussolati B, Omede P. et al. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia. 2012;26:1081-90
34. De Rossi G, Evans AR, Kay E, Woodfin A, McKay TR, Nourshargh S. et al. Shed syndecan-2 inhibits angiogenesis. J Cell Sci. 2014;127:4788-99
35. Yan Z, Liu J, Xie L, Liu X, Zeng Y. Role of heparan sulfate in mediating CXCL8-induced endothelial cell migration. PeerJ. 2016;4:e1669
36. Dyer DP, Salanga CL, Volkman BF, Kawamura T, Handel TM. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology. 2016;26:312-26
37. Taleb S. Inflammation in atherosclerosis. Arch Cardiovasc Dis. 2016
38. McAlpine CS, Swirski FK. Circadian Influence on Metabolism and Inflammation in Atherosclerosis. Circulation research. 2016;119:131-41
39. Sorci-Thomas MG, Thomas MJ. Microdomains, Inflammation, and Atherosclerosis. Circulation research. 2016;118:679-91
40. Yurdagul A Jr, Finney AC, Woolard MD, Orr AW. The arterial microenvironment: the where and why of atherosclerosis. Biochem J. 2016;473:1281-95
41. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:2191-8
42. Simmons RD, Kumar S, Jo H. The role of endothelial mechanosensitive genes in atherosclerosis and omics approaches. Archives of biochemistry and biophysics. 2016;591:111-31
43. Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta physiologica (Oxford, England). 2016
44. Brown AJ, Teng Z, Evans PC, Gillard JH, Samady H, Bennett MR. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat Rev Cardiol. 2016;13:210-20
45. Aluganti Narasimhulu C, Fernandez-Ruiz I, Selvarajan K, Jiang X, Sengupta B, Riad A. et al. Atherosclerosis - do we know enough already to prevent it? Curr Opin Pharmacol. 2016;27:92-102
46. Wang Y, Qiu J, Luo S, Xie X, Zheng Y, Zhang K. et al. High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater. 2016;3:257-67
47. Wu GF, Du ZM, Hu CH, Zheng ZS, Zhan CY, Ma H. et al. Microvessel angiogenesis: a possible cardioprotective mechanism of external counterpulsation for canine myocardial infarction. Chinese medical journal. 2005;118:1182-9
48. Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G. et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1664-8
49. Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res. 2016;119:375-96
50. Dumont O, Loufrani L, Henrion D. Key role of the NO-pathway and matrix metalloprotease-9 in high blood flow-induced remodeling of rat resistance arteries. Arterioscler Thromb Vasc Biol. 2007;27:317-24
51. Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y. et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science (New York, NY). 2007;316:295-8
52. Choi S, Snider AJ. Sphingolipids in High Fat Diet and Obesity-Related Diseases. Mediators Inflamm. 2015;2015:520618
53. Meyer zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochimica et biophysica acta. 2007;1768:923-40
54. Kimura T, Watanabe T, Sato K, Kon J, Tomura H, Tamama K. et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem J. 2000;348(Pt 1):71-6
55. Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP. et al. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367-73
56. Curry FR, Adamson RH. Tonic regulation of vascular permeability. Acta physiologica (Oxford, England). 2013;207:628-49
57. Poti F, Gualtieri F, Sacchi S, Weissen-Plenz G, Varga G, Brodde M. et al. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R-/- mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1505-12
58. Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A. et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. 2012;23:600-10
59. Wang HW, Liu PY, Oyama N, Rikitake Y, Kitamoto S, Gitlin J. et al. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR-/- mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3561-70
60. Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M. et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301-12
61. Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1312-8
62. Kim GS, Yang L, Zhang G, Zhao H, Selim M, McCullough LD. et al. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun. 2015;6:7893
63. Skoura A, Michaud J, Im DS, Thangada S, Xiong Y, Smith JD. et al. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:81-5
64. Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7988-95
65. Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a "bumper-car" model. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16483-8
66. Ebong E, Lopez-Quintero S, Rizzo V, Spray D, Tarbell J. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integrative biology: quantitative biosciences from nano to macro. 2014
67. Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136-42
68. Yen WY, Cai B, Zeng M, Tarbell JM, Fu BM. Quantification of the endothelial surface glycocalyx on rat and mouse blood vessels. Microvascular research. 2012;83:337-46
69. Yao Y, Rabodzey A, Dewey CF Jr. Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. 2007;293:H1023-30
70. Lipowsky HH. Protease Activity and the Role of the Endothelial Glycocalyx in Inflammation. Drug Discov Today Dis Models. 2011;8:57-62
71. Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. 2014;306:H363-72
72. Zhang L, Zeng M, Fan J, Tarbell JM, Curry FR, Fu BM. Sphingosine-1-phosphate Maintains Normal Vascular Permeability by Preserving Endothelial Surface Glycocalyx in Intact Microvessels. Microcirculation. 2016;23:301-10
73. Zeng Y, Liu XH, Tarbell J, Fu B. Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Exp Cell Res. 2015;339:90-5
74. Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS. et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8:693-704
75. Adyshev DM, Moldobaeva NK, Elangovan VR, Garcia JG, Dudek SM. Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal. 2011;23:2086-96
76. Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X. et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692-700
77. Surya VN, Michalaki E, Huang EY, Fuller GG, Dunn AR. Sphingosine 1-phosphate receptor 1 regulates the directional migration of lymphatic endothelial cells in response to fluid shear stress. Journal of the Royal Society, Interface. 2016;13:pii 20160823
78. Dela Paz NG, Melchior B, Frangos JA. Shear stress induces Galphaq/11 activation independent of G protein-coupled receptor activation in endothelial cells. American journal of physiology Cell physiology. 2017 ajpcell.00148.2016
79. dela Paz NG, Melchior B, Shayo FY, Frangos JA. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Galphaq/11 subunits of heterotrimeric G proteins. J Biol Chem. 2014;289:7413-24
Author contact
![]() Corresponding author: Ye Zeng, yeedu.cn Institute of Biomedical Engineering, West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, No.17, Section 3, Renmin South Road, Chengdu, Sichuan 610041, P.R. China Tel/Fax: +86-028-85502314
Corresponding author: Ye Zeng, yeedu.cn Institute of Biomedical Engineering, West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, No.17, Section 3, Renmin South Road, Chengdu, Sichuan 610041, P.R. China Tel/Fax: +86-028-85502314